Now, you can view who all are reading this site live on a revolving globe. It makes all the more happier to note that all these grateful and honourable dots (i.e., you) are literally drawing the world map . Six million reads from 190 countries, right from the Solomon Islands in Micronesian Pacific, abutting the International date line, to the extreme west, reaching Chile and Hawaii has happened so far.
This is a 15-year-old post about LVH, written in 2008. Few of my colleagues, now agree with this, but still hesitate to oblige in the open, suggesting it is too good to be true! Re-posting it for your own assessment. Surprised, why cardiology community didn’t consider this observation worthy to pursue.
Advantages of Left ventricular hypertrophy (LVH)
Left ventricular hypertrophy is one of the most common clinical cardiac entity.It is recognised either by ECG or echocardiography.LVH has a unique place in cardiology as it can imply a grossly pathological state or a marker of healthy heart as in physiological hypertrophy in athletes.
Logic would suggest, in this era of stem cells and nano medicine , every muscle fibre in ventricle is worth in gold !. So when the nature provides an extra reserve of myocardium in the form of LVH one should welcome it, if otherwise not harmful.
Is LVH due to systemic hypertension benign ?
Not really, LVH has been shown to be an independent cardiac risk factor. (The famous Framingham study)Further LVH can result in diastolic dysfunction and the risk of cardiac failure increases.
But in spite of these observations, an astute clinician with considerable experience will appreciate , patients with LVH fare better during an acute coronary syndrome !
This has been a consistent clinical observation . (Shall we call it as class C . ACC /AHA evidence? )
Is LVH an asset during ACS ?
A hypertrophied heart takes ischemic injury very easy , it doesn’t really hurt much . Another possibility is that in LVH myocytes are relatively resistant to hypoxia .
Patients with LVH rarely show significant wall motion defect following an STEMI.This is probably because the full thickness transmural necrosis is almost never possible even if extensive MI occurs.
This is also reflected in ECG as these patients rarely develop q waves in following STEMI .
Persistent ST elevation and failed thrombolysis is very uncommon in pateints with LVH.
LVH provides a relative immunity against development of cardiogenic shock . It requires 40% of LV mass destruction to produce cardiogenic shock.This can rarely happen in LVH. In a long term analysis we have found none of the patient with LVH developed cardiogenic shock following STEMI.
LVH patients are also protected against development of free wall rupture.
Concluding message
“Lack of published evidence is the weakest evidence to dismiss a true myth”LVH , either pathological or physiological, has a hitherto unreported beneficial effect.It acts as a myocardial reserve and helps limit the impact of STEMI.
Pardon ,this video is nothing to do with cardiology. It is from the archives of the United nations library .This can teach some important lessons in art of communication , sharing to all folks, especially medical students. It was recorded in 1959 in Newyork, UN head quarters.Four 17 year old school girls & boys were invited for a debate on a complex topic. Does God exist ? How do you pray ? and what is the purpose of different religions ?
I keep wondering , how these youngsters accumulated so much wisdom and express it in such a polite manner too. Mind you, this was recorded , when learning happened with out any digital aids.The word Internet was unheard off. No ego, no bluntness, no diatirbes that has become a norm in many debates now. I got a punching lesson from this clip, be gentle when taking extreme views in any topic.
I wish, every medical debate in class rooms should happen this way.The key to succesful debate is, to accumulate knowedge, willingness to question the convention, and respecting the oppositie point of view.
The high point of talk show, was, when the Brazilian girl(due respects, she should be nearing 80 years now) tell us casually some things are not meant to be understood in life .I tell the same when some patients ask too many questions about their illness which may not have an answer.
Wishing every one of you an Enlightening New year. As we begin a new journey around the sun, yet another time, let us re-dedicate ourself, to use science, for the welfare of our planet & people.
Thank you , for visiting this site and make all its worth.
Just one memory of 2022, lingers ! Retired and left Madras medical college,Chennai after 3 decades, which grew me up as a Cardiologist.
“I thought, he was not the right patient for the procedure. I believe, what I did was the correct decision. Why all this fuzz? after all, the patient is doing so well without that procedure,.. are you worried about that?
“No, I need an explanation, we have a fully functional cath lab in our center. The patient came in the right window period. Still, you haven’t offered the best mode of treatment”.
“I can reiterate it again sir. Just because a lab is available 24/7, it doesn’t make all patients eligible for a PCI. I think I didn’t commit a professional misdemeanor when I decided in favor of fibrinolysis. In fact, I would be guilty had I rushed him to the cath lab, just to satisfy the misplaced scientific position we have decided to adopt. If you think, I am culpable for successfully treating a patient without taking the patient to the cath lab, you may proceed with the penal action.
Before that, I would request you to please read the current edition of this book we all revere. (Which continues to mentor physicians all over the globe for the past 50 years)
The current edition of Harrison 2022 is just out. I thought, there is something great learning point in Cardiology chapter, specifically about the reperfusion strategies in STEMI
My hearty thanks to the editors of the chapter for the crystal clear expression about this much-debated procedure* and specifically choosing the word “PCI appears* to be more effective ” (even) if it is done in experienced persons in dedicated centers. The choice of the word used by the authors is Intentional and must be applauded. This message must be propagated to all our fellow physicians. What a way to convey an important truth pertaining to the management of the most common cardiac emergency, while many in the elite specialty are so dogmatic in their assertion without verifying the reality.
* The verdict is still under the jury even after 3 decades, since the PAMI days of the early 1990s. Thank you, Harrison. What a gentle, but a righteous way to express an opinion about a procedure that is apparently enjoying a larger-than-life image based on a handful of studies and a flawed meta-analysis.
Final message
Primary PCI is just an alternateform of treatment to fibrinolysis in STEMI. Both are equipoise in the majority of patients. Extreme care and diligence are required to harvest the small benefit the PCI seems to provide. There are lots of ” if and buts” that decide the success of this procedure. Get trained, and do it selectively for those who really need it.
Postamble
You may call yourself a super-specialist. But, please realize, If you have any doubt about key management strategies, never feel shy to take a cue from Internal medicine books. The greatness of these warrior books is that, it comes devoid of all those scientific clutters backed by premature evidence.
Dr.Richard Asher, a British physician from Sussex addressed a group of young passing out medical students way back in 1948 in London. The lecture was titled seven sins of medicine! We should thank the Lancet for having published this brief speech the subsequent year in its journal making it immortal medical teaching!
Though he was listing these sins among medical students, it is very relevant to every health professional.
1.Obscurity
Asher endorses the use of clear communication and plain language whether writing or speaking. Obscurity may be used to cloak one’s own ignorance, or due to an inability to communicate with those outside of the medical profession. “If you don’t know, don’t admit it. Instead, try to confuse your listeners.” is not uncommon. Regardless of the intention, whether to misdirect from incompetence or to foster a feeling of superiority, the patient and those surrounding them are often left confused and uncertainiy.
2. Cruelty
This sin is perhaps one of the most commonly committedby doctors and medical students. Whether it be the physical thoughtlessness of a half-dozen students palpating a painful tumor mass, or loudly taking (or presenting) a patient’s history in a crowded room, one of the first things that is unlearnt by a medical professional is to treat the patient as they themselves would like to be treated.
3.Bad Manners
Often overlooked, rudeness or poor taste in humour is condoned within the hospital setting. At the end of the day, many doctors and students are simply rude to patients that do not suit them. Whether it is a snapping at an uncooperative patient or making a cruel joke about them after leaving the room, the impact of these “coping mechanisms” (as they are considered to be by many) must be taken into account.
4. Over-Specialisation
In a growing trend by the medical establishment, over-specialization and under-generalization is a growing problem in the wider medical community. Ignoring aspects of one’s education in favor of more interesting aspects is a behavior that is pathological and outright negligent in a student. Failure to diagnose or to treat a patient because “their signs and differential fall outside of my field, let’s turf them to another service” ought to be a seriously considered Supervisory & Training issue.
5.Love of the Rare
(aka “If you hear hoof-beats, think horses. Not zebras”) The desire for rare and interesting diseases causes many medical students and young doctors to seek the bizarre rather than seeing a mundane diagnosis.
6. Common Stupidity
As well as the standard definition for this sin, the specific example of “using empirical procedures rather than tailoring for the patient” or the young doctor “flying on autopilot” must be mentioned. Ordering another test that is redundant, and for which the results may already be interpreted from the history, before starting treatment is such a situation. For example: requesting a hemoglobin count before beginning transfusion, despite the fact that the patient appears obviously anaemic.
7. Sloth
Laziness. Also includes ordering excessive numbers of tests, rather than simply taking the time to take an adequate history
Final message
It is astonishing, to note Dr.Asher made this observation in the very early days in the evolution of modern medicine,(No critical care units, no HMOs, No industry nexus with research, & commodification of medicine ) I wonder what Dr. Asher would have to write if he is alive in 2021.
Wish, every medical professional shall find their Asher score. Looking back on my career, I must confess my score would be 3 ( may be 3.5 !) out of 7. Now, desperately trying to get rid of them. Mind you, the 4th (Overspecailisation) and 6 th (common stupidity) is inherently built into the system. I think, very tough to avoid them.
I haven’t clearly understood the true meaning of customary Dr tag, my name carries for more than 3 decades, till I saw this. Wish, this video is played to all young medical students on their graduation day.
I am realizing with guilt, it requires a Holywood movie buff to remind us the true meaning of the famous WHO – definition of Health, done in the most holistic fashion in the year 1948.
Health is a state of complete physical, mental, and social well-being and not merely the absence of disease or infirmity.
So, technically, whoever serves to improve these three components and alleviate human suffering becomes a doctor.
Happy to share this on July 1st, the official Doctor’s day in India in memory of the Bharat Ratna Dr.B.C.Roy of Bengal.
Reference
The clip is from the movie Patch Adams, Directed by Tom Shadyac. A Hollywood celebrity movie maker, Virginian professor of communication turned philanthropist, now retired to a minimalist life. He is also known for his famous documentary I amthat talks about the problems faced by the world. Though his works are much appreciated, I must say, they are underrated. Deserves more than an Oscar for communicating his thoughts on the medical profession perfectly and for social equality.
I think it is an Invalid question. Whether you like it or not , medical science and philosophy are always bonded together and its relationship is eternal. It doesn’t make sense to separate them. I think we have misunderstood the meaning of philosophy. While science is presumed truths, philosophy is trying to believe in unknown truths. Philosophical truths are built-into every decision a medical professional takes.
If the expected natural history of any disease is science, unexpected deviations are philosophy. (RT PCR testing for diagnosing Corona is science, why 90% of them are not infective and don’t transform disease is philosophy) When something is not seen or quantifiable like human immunity, it is a perfect example of concealed science or manifest philosophy.
Taking about what we think we know is science, Talking about what we really don’t know is philosophy. The term Idiopathic syndrome finds a proud of the place in every specialty in medicine, Isn’t?
What will be your answer when your patient wants an assurance that a stent, you had just implanted will not get occluded in the next 6 months or so.“I don’t know, I cant assure you about that” will be your most likely answer. (Though, we do it in style, hiding behind the scientific hyperbole decorated with numbers, also referred to as statistics) Please realize, this is the expression of medical philosophy in the finest form.
Final message
My Impression is, philosophical truths should be liberally used in a regular fashion right from the first-year medical school to advanced specialty teaching. This seems essential as science in the current times suffers from too much sanctity. This has spilled over to the doctor population as well, and make them appear invincible.
If only we realize science often trailsbehind the philosophical truths at least by a few decades, our patients will not be injured inappropriately and prematurely. Mixing science with philosophy in the right composition ( aperfect academic cocktail ) will bring out the best from the noble profession.
Postamble
Can anyone guess, why scientists are given a doctorate in Philosophy degree (PhD ) ?
A young man aged around 40 years, had a STEMI was promptly thrombolysed in a small hospital located about 40 KM away in the suburbs of my city Chennai. They did an awesome job of saving the patient life and salvaging the myocardium.
Now begins the story . . . one of the non-medical person who is the owner of the hospital has an unfortunate working business relationship with a frighteningly big nearby hospital which had signed a memorandum of irresponsible understanding . It demanded any patient who arrives in the small hospital with MI should be transferred at earliest opportunity to them.
So, an ambulance was arranged and the patient (with a fairly well reperfused heart ) was shifted in an emergency fashion . It reached desired destination after nicely chugging along the choked chaotic Chennai evening traffic for 45 minutes.
The guy was taken directly to cath lab through the side doors to perform a second salvage procedure on a successfully opened IRA. Young cardiology consultants in designer cath suite welcomed the smiling ACS patient to their posh new lab .Did few rapid radial shots, mumbled among themselves for few minutes, decided to stent a minimal LAD lesion for a patient who was in zero distress with well-preserved LV function.
*The relatives of the patients were curious when they were asked sign a fresh set of consent which elaborately mentioned about possible life risk during the procedure.
The patient’s wife was clearly amused and she pointed out to the superior cardiologists about the earlier briefing by the Inferior freelance cardiologist who treated him in the previous hospital. She recalled , “I was told in confident terms that Initial thrombolysis has been spectacularly successful and bulk of the treatment is over and risk of complication has dramatically reduced”.
Then why is this distressing risk taking story again , she asked ?
The doctors hurriedly explained ,”this procedure is different. We are sorry to say we have no other option but to add further risk to you” ! but , its all for your good !
Why should I ? If the initial lysis is very successful why do you want to meddle with it again ?
No Madam , you are ill-informed , you can’t talk like that .This is what modern science is all about. Leave the professional decision to us. We need to check immediately whether the lysis is really successful .We can’t rely on the ECG.Further, true success lies in stenting the lesion as we fear the ill-fated site may close again.We are taught to practice protocols based on standard scientific guidelines. This hospital has highest rating in-terms of quality care. That’s why we got updated ISO 2000 NABH accreditation
The women who is a soft ware engineer was smartly and scientifically silenced in 5 minutes flat !
Post-amble :
What happened to the patient then ? (When you fear something it happens is in’t the Murphy’s law ?)
The apparently asymptotic and comfortable patient had uneventful PCI. A long drug eluting stent was implanted in recanalized lesion in LAD with around 30 % narrowing that ended with an innocuous looking diagonal pinch. The procedure was uneventful , however next day he developed some fresh ECG changes and chest pain . The worried team took him for another angio found stent was patent But , ultimately after a stressful 3 days of stay , some thing went wrong he ended up with new LV dysfunction.He got discharged fine with a caution that , his stent needs to intensively monitored for the next 1 year since technically he had recurrent ACS !
Lessons we don’t learn from such cases.
When two procedures are done to accomplish the same aim (Reperfusion) , but with differing success rates, expertise, time ,and unpredictable hazards , the benefits from them may not add together. There is clear knowledge deficit here. Scientific data can never provide fair answers to these questions as all real life cofounders can never be recreated in study population.
While we expect 1+1 to become two in pharmaco-Invasvie strategy ,one should realise it may end up with either zero or even – 2 .
1 -1 = 0
-1 + (-1)= -2 ?
Learning cardiology from lay persons
The patient’s shrewd wife threw this question ,
After two modes of re-perfusion done sequentially in my husband’s heart , at a total cost of Rs4.5Lakhs Why he is still left with significant LV dysfunction (Which was around 40% EF.)
The query raised by the lady appeared much more crucial and logical than the ones discussed in many top-notch live interventional workshops we attend every few months!
As usual , I started mulling over the issue. There is something wrong with the way , we understand the pharmaco invasive approach-PIA .You go with it only if initial pharmacological approach has failed.
Of Course ,there is one more modality possible ie Pharmaco -Angio strategywhere in, you look at the coronary anatomy and take a call ! This sounds good , the only issue is taking a right call! My experience suggests wrong calls are the rule and exceptions are rare. Then a whole new issue erupts about all those non IRA lesions
Final message
So, til we have gain complete self-control over our evolved ignorance and evolving knowledge , it is better to follow this proposed funny new ACS algorithm called “Pharmaco -non invasive” approach (PNIA) in asymptomatic ACS patients who have had apparently successful lysis.
*Please note, Incidentally PNIA actually refers to simple good old traditional stand alone thrombolysis.
Counter point
No one can deny Interventional cardiology carries a risk of untoward effects.Don’t blow this out of proportion. Do you know, how many lives have been saved by routine Pharmaco -Invasive approach ?
I am not sure , my experience may be limited.Let me ask the readers. Is routine PIA is warranted in all asymptomatic , successfully lysed STEMIs ?
100% occlusion of a coronary artery result in STEMI.This includes both thrombus and mechanical component .We are very much blinded till we touch , feel and see the lesion with a wire or IVUS to quantify the mechanical component’s contribution in the genesis of STEMI.It is generally believed (True as well ) thrombus is the chief culprit .It can even be 100 % thrombotic STEMI with just a residual endothelial erosion and hence
zero mechanical component .However , the point of contention that non flow limiting lesion is more likely to cause a thrombotic STEMI than a flow liming
lesion seems to be biased and misunderstood scientific fact .
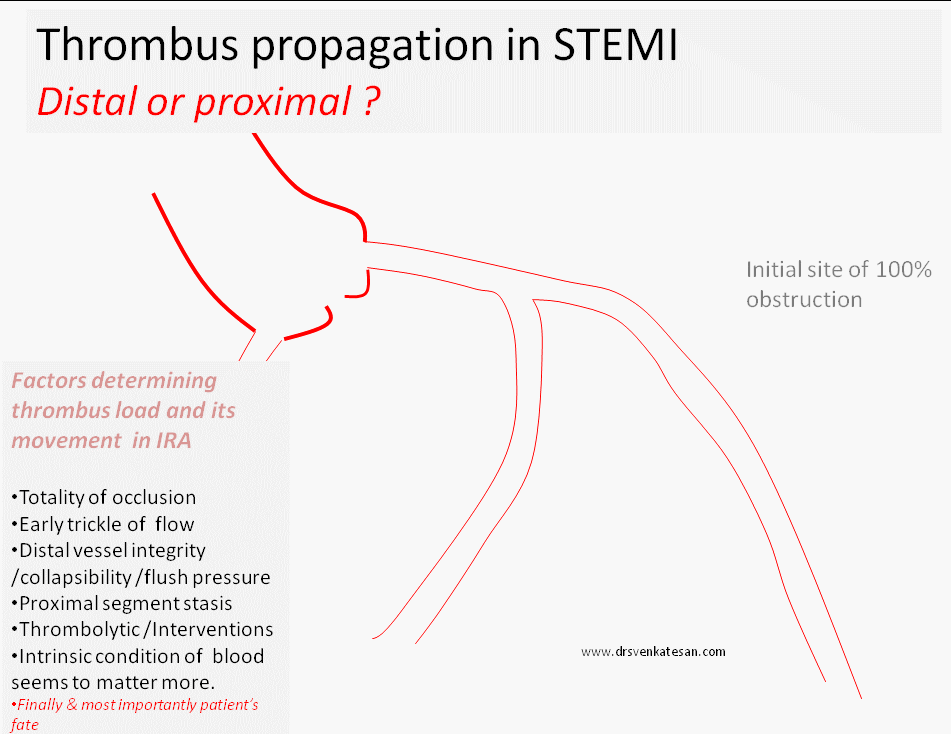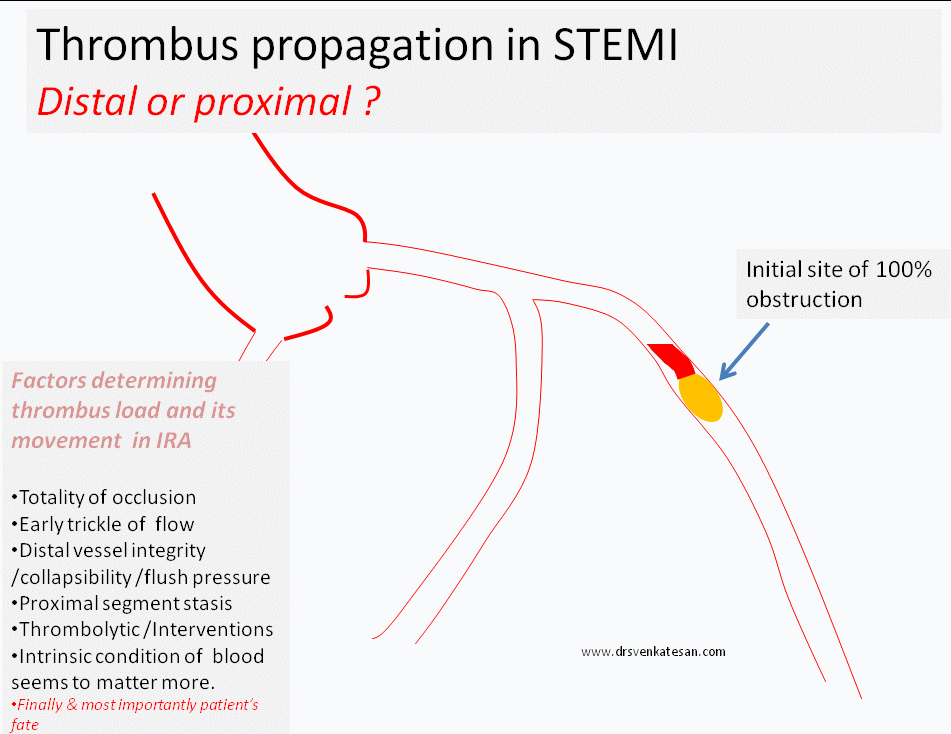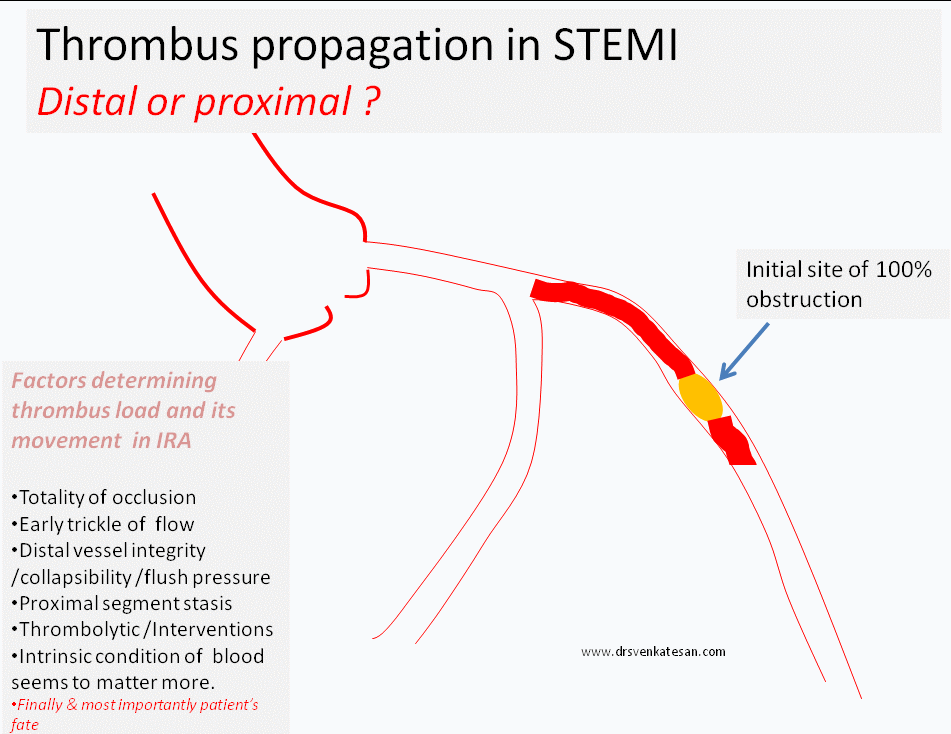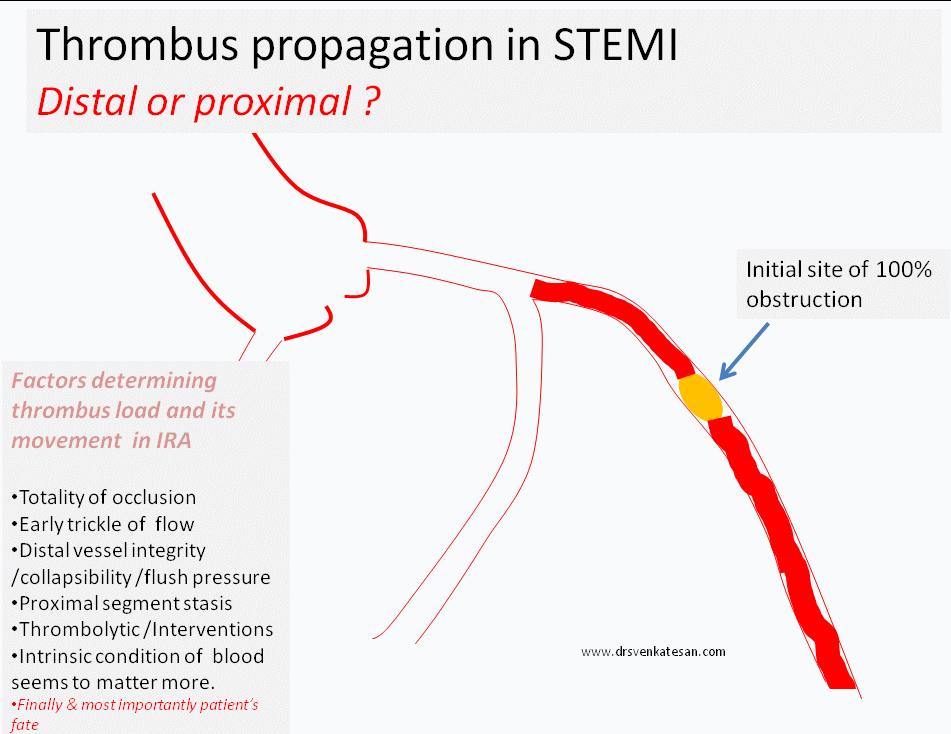
What happens once 100 % occlusion take place ?
Sudden occlusion , is expected to evoke a strong fire fighting response within the coronary artery.The immediate reaction is the activation of tissue plasminogen system. In this aftermath few succumb . ( Re-perfusion arrhythmia generated as VF ) .The TPA system activates and tries to lyse the clot.The volume , morphology, attachment, content of thrombus , and the elasticity of fibrin mesh , location of platelet core would determine the life and dissolvablity of thrombus. Even a trickle flow can keep the distal vessel patent .(Please note a timely TIMI 2 flow can be a greater achievement than a delayed TIMI 3 flow !)
What happens to the natural history of thrombus in STEMI ?
Thrombus formed over the culprit lesion can follow any of the following course
Can remain static
Get lysed by natural or pharmacological means
Progress distally (By fragmentation or by moving en-mass )
Grow proximal and and involve more serious proximal side branch obstruction
Organise and become a CTO
Factors determining thrombus migration
The interaction between the hemodynamic forces that push a thrombus distally and hemo-rheological factors that promote fresh proximal thrombus formation are poorly understood. The altered intra-coronary milieu with a fissured plaque covered by platelet vs RBC / fibrin core, totally of obstruction, reperfusing forces , re-exposure of raw areas and the distal vessel integrity all matters.
While, logic would tell us, thrombus more often migrates distally assisted by the direction of blood flow, an opposite concept also seeks attention , ie since the blood flow is sluggish in the proximal (to obstruction site )more thrombus forms in segments proximal to obstruction.
(In fact, its presumed in any acute massive proximal LAD STEMI , it takes hardly few minutes for the thrombus to queue up proximaly and clog the bifurcation and spill over to LCX or even reach left main and result in instant mechanical death.)
What is the significance of length and longitudinal resistance of the thrombotic segment in STEMI ?
If thrombus is the culprit let us get rid of it , this concept looks nice on paper , but still we don’t know why thrombus aspiration in STEMI is not consistently useful. We also know little about the length of the thrombotic segment .When a guide wire is passed over a STEMI ATO it may cross smoothly like “cutting a slice of butter” in some , while in few we struggle and end up with severe no-reflow inspite of great efforts .Why ?
What is the Impact of distal collateral flow in flushing fresh thrombus ?
The efficacy of collateral flow in salvaging myocardium is underestimated. Distal vessel flow if perfused partially by acute collaterals the thrombus load is not only less it’s soft and fail to get organised early that would help cross the lesion easily.
Mohandas Karam Chand Gandhi , father of my country , India , made these observations in year 1925 about the fundamental constituents of violence in society . These words of monumental wisdom came when he was addressing young Indians in a country- side rally .
Note, his finger points to , what exactly is relevant to our profession ! He emphasized this nearly 100 years ago, when medical science was at its infancy .One can only guess what would be Mahatma’s comment about our profession in it’s current form !
Should we include moral, behavioral and ethical classes right from the first year of medical school along with Anatomy , physiology and bio chemistry.Medical council of India obviously need to burn more mid night oil , I wish it happens in my life time. !
I don’t know, any one has tried to differentiate the mechansims of dyspnea with reference to systolic and diastolic dysfunction .We have made some observations in certain group of patients during EST . I do not know how far one would agree with this .
For the same amount of stress or work load persons with systolic dysfunction behave differently . However ,both will complete the activity but the onset and perception of dyspnea is slightly different in patients with predominant diastolic dysfunction.
Diastolic dyspnea (Dyspnea due to predominant diastolic dysfunction / HFPEF)
Delayed dyspnea . It manifest well after the exertion is completed.
It is more off a struggle to handle the venous return .The forward flow (Arterial circuit ) is relatively well toned and tuned and hence fatigue is rare .
Typically it has a prolonged recovery time .(? > 1-2 minutes )
Is it less harmful in terms of longevity ? May be . . . since it is more related to physical de-conditioning. Most of the physiological episodes of dyspnea are probably diastolic dysfunction mediated .
Dyspnea that is triggered in diastole is also dependent very much on the heart rate .If the heart rate fail to reach the baseline the recovery of dyspnea is also delayed
Some believe , physiological dyspnea should disappear within 30-60 seconds after termination of activity .(Highly arbitrary!)
Systolic dyspnea (Dyspnea due to predominant systolic dysfunction )
Patients with primary systolic pump failure experience dyspnea very early into exercise .
Much of dyspnea occur during activity itself .
Exercising muscles show hypoxia and hence fatigue is conspicuous .
Recovery of dyspnea is relatively immediate as the activity is stopped .Demand from exercising muscle is significantly dropped.
If the venous return is well handled by the ventricles the recovery phase is more comfortable .
Summary
In primary diastolic dysfunction ,the maximum stress to ventricle occurs when the venous return peaks that usually happen in the exercising muscles , as they shed vaso-dilatory property in post exertion phase .
Management Implication
Fluid overload , Tachycardia are more related to diastolic dysfunction .(Beta blockers by prolonging the diastole can , provide important relief of dyspnea in diastolic dysfunction (In HOCM patients this action could be more important that the much hyped negative inotropism !)
Final message
Dyspnea is a complex cortical perception , influenced by filling pressure of heart, stretch receptor in lungs , respiratory and exercise muscle . It is further impacted by number of biochemical parameters (Lactate/ O2 etc )
Of-course , it could be a far fetched imagination to split dyspnea mechanism with reference to cardiac cycle. Combinations of both systolic and diastolic dysfunction is the norm in many cardiac conditions . However , I believe we need more insight in the pathogenesis of this , “most important symptom” that emanate from the heart .
Prosthetic valve implantation has revolutionized the management of valvular heart disease . The original concept valve was a ball in a cage valve , still considered as a fascinating discovery. It was conceived by the young Dr Starr and made by Engineer Edwards .This was followed by long hours of arguments, debates and experiments that ran into many months . The silent corridors of Oregon hospital Portland USA remain the only witness to their hard work and motivation. At last, it happened , the first human valve was implanted in the year 1960. Since then . . . for nearly 50 years these valves have done a seminal job for the mankind.
With the advent of disc valve and bi-leaflet valve in the later decades of 20th century , we had to say a reluctant good-bye to this valve.
There is a lingering question among many of the current generation cardiologists and surgeons why this valve became extinct ?
Starr and Edwards with their child !
We in India , are witnessing these old warrior inside the heart functioning for more than 30 years.From my institute of Madras medical college which probably has inserted more Starr Edwards valve than any other during the 1970s and 80s by Prof . Sadasivan , Solomon victor , and Vasudevan and others .
It is still a mystery why this valve lost its popularity and ultimately died a premature death.The modern hemodynamic men working from a theoretical labs thought this valve was hemodynamically inferior. These Inferior valves worked like a power horse inside the hearts the poor Indian laborers for over 30 years.
A Starr Edwards valve rocking inside the heart in mitral position
The cage which gives a radial support* mimic sub valvular apparatus, which none of the other valves can provide.
* Mitral apparatus has 5 major components. Annulus, leaflets, chordae, pap muscle, LV free wall.None of the artificial valves has all these components. Though , we would love to have all of them technically it is simply not possible. The metal cage of Starr Edwards valve partially satisfies this , as it acts as a virtual sub valvular apparatus.Even though the cage has no contact with LV free wall, the mechano hydrolic transduction of LV forces to the annulus is possible .
Further , the good hemodyanmics of this valve indicate , the cage ensures co axial blood flow across the mitral inflow throughout diastole. .Unlike the bi-leaflet valve , where the direction of blood flow is determined by the quantum of leaflet excursion in every beat . In bileaflet valves each leaflet has independent determinants of valve motion . In Starr Edwards valve the ball is the leaflet . In contrast to bi-leaflet valve , the contact area of the ball and the blood in Starr Edwards is a smooth affair and ball makes sure the LV forces are equally transmitted to it’s surface .
The superiority of bi-leaflet valves and disc valves (Over ball and cage ) were never proven convincingly in a randomized fashion . The other factor which pulled down this valve’s popularity was the supposedly high profile nature of this valve. LVOT tend to get narrowed in few undersized hearts. This can not be an excuse , as no consistent efforts were made to miniaturize this valve which is distinctly possible.
Sudden deaths from Starr Edwards valve .
Almost unheard in our population.
The major reason for the long durability of this valve is due to the lack of any metallic moving points .
Absence of hinge in this valve confers a huge mechanical advantage with no stress points.
A globe / or a ball has the universal hemodynamic advantage. This shape makes it difficult for thrombotic focus to stick and grow.
Final message
Science is considered as sacred as our religion . Patients believe in us. We believe in science. A good durable valve was dumped from this world for no good reason. If commerce is the the main issue ( as many still believe it to be ! ) history will never forgive those people who were behind the murder of this innocent device.
Cardiologists and Cardio thoracic surgeons are equally culpable for the pre- mature exit of this valve from human domain. Why didn’t they protest ? We can get some solace , if only we can impress upon the current valve manufacturers to give a fresh lease of life to this valve .
This was written originally in 2009 early days of this blog. Now, re-posting it in 2021 , wonder any one has new data on this!
We know diabetes, smoking, hyperlidemia, hypertension are major risk factors for progressive vascular disease. They damage the vascular endothelium either directly or indirectly , by aggravating the atheroscelortic process . Diabetes apart from affecting the medium sized arteries , also affect the microvasculature. Smoking has a direct effect on endothelial function .It depletes vascular nitric oxide. High levels of circulating lipids injures the sub endothelial structures and invades the media by entering macrophages .So , all these 4 risk factors either operate independently or interact with each other and result in progressive vascular disease.
While we believe , these risk factors do not have any bias in attacking the human vascular tree, in the real world it is observed they have their own behavior pattern and have unique predilection and a deadly alliance .
For example , in chronic smokers TAO is the commonest manifestation , thrombo angitis is far too less common to occur in the coronary arteries.
Similarly hypertension per se rarely results in an acute coronary syndrome while it is the single important cause for cerebro vascular disease. Diabetes especially in women has very strong predilection for CAD , while diabetic per se is a lesser risk for stroke. Hyperlipedimia may be the one which has fairly even risk throughout the vasculature. Similarly there is a difference in renal and carotid arterial involvement with reference to the conventional risk factors .
Why this apparent difference ?
We are unlikely to get an answer to this question in the near future . Left to the youngsters . . . of tomorrow !
* Note of clarification
The source for the above chart is collected from various studies and also a huge observational data from our hospital. There could be some geographical variation , a given individual may respond differently to these risk factor depending upon his genetic predisposition and susceptibility . So the above data can be applied to general population and not to a individual.
It is often said life is a cycle , time machine rolls without rest and reach the same point again and again . This is applicable for the knowledge cycle as well .
We live a life , which is infact a “fraction of a time”(<100years) when we consider the evolution of life in our planet for over 4 million years.
Man has survived and succumbed to various natural and self inflicted diseases & disasters. Currently, in this brief phase of life , CAD is the major epidemic , that confronts modern man.It determines the ultimate life expectancy . The fact that , CAD is a new age disease and it was not this rampant , in our ancestors is well known .The disease has evolved with man’s pursuit for knowledge and wealth.
A simple example of how the management of CAD over 50 years will help assess the importance of “Time in medical therapeutics”
1960s: Life style modification and Medical therapy is the standard of care in all stable chronic CAD The fact is medical and lifestyle management remained the only choice in this period as other options were not available. (Absence of choice was a blessing as we subsequently realised ! read further )
The medical world started looking for options to manage CAD.
1970s : CABG was a major innovation for limiting angina .
1980s: Plain balloon angioplasty a revolution in the management of CAD.
1990s: Stent scaffolding of the coronaries was a great add on .Stent was too dangerous for routine use was to be used only in bail out situations
Mid 1990s : Stents reduced restenosis. Stents are the greatest revolution for CAD management.Avoiding stent in a PCI is unethical , stents should be liberally used. Every PCI should be followed by stent.
Stents have potential complication so a good luminal dilatation with stent like result (SLR) was preferred so that we can avoid stent related complications.
2000s: Simple bare metal stents are not enough .It also has significant restenosis.
2002: BMS are too notorius for restenosis and may be dangerous to use
2004 : Drug eluting stents are god’s gift to mankind.It eliminates restenosis by 100% .
2006: Drug eluting stents not only eliminates restenosis it eliminates many patients suddenly by subacute stent thrombosis
2007 : The drug is not the culprit in DES it is the non bio erodable polymer that causes stent thrombosis. Polymer free DES or biodegradable stent , for temporary scaffolding of the coronary artery (Poly lactic acid ) are likely to be the standard of care .
All stents are potentially dangerous for the simple reason any metal within the coronary artery has a potential for acute occlusion.In chronic CAD it is not at all necessary to open the occluded coronary arteries , unless CAD is severely symptomatic in spite of best medical therapy.
2007: Medical management is superior to PCI in most of the situations in chronic CAD .(COURAGE study ) .Avoid PCI whenever possible.
2009 :The fundamental principle of CAD management remain unaltered. Life style modification, regular exercise , risk factor reduction, optimal doses of anti anginal drug, statins and aspirin is the time tested recipe for effective management of CAD .
So the CAD therapeutic journey found it’s true destination , where it started in 1960s.
Final message
Every new option of therapy must be tested against every past option .There are other reverse cycles in cardiology that includes the role of diuretics in SHT , beta blockers in CHF etc. It is ironical , we are in the era of rediscovering common sense with sophisticated research methodology .What our ancestors know centuries ago , is perceived to be great scientific breakthroughs . It takes a pan continental , triple blinded randomised trial to prove physical activity is good for the heart .(INTERHEART , MONICA studies etc) .
Medical profession is bound to experience hard times in the decades to come , unless we look back in time and “constantly scrutinize” the so called scientific breakthroughs and look for genuine treasures for a great future !
Common sense protects more humans than modern science and it comes free of cost too . . .
Exercise stress test ( Also called treadmill test ) is an important investigation not only in patients with suspected CAD but also in established CAD . In the former group , it helps us to exclude CAD in patients with chest pain and in the later group , it helps us to assess functional capacity , risk stratification and to detect any additional ( New or residual ) ischemia.
Stress test being a physiological test , has a huge advantage of assessing the adequacy of myocardial blood flow without even knowing the coronary anatomy , while Coronary angiogram (CAG) has a zero physiological value* in spite of excellent assessment of the coronary anatomy !
It is an irony , in the assessment of angina we are expected to assess the physiological adequacy of myocardial blood flow , we have kept coronary angiogram as a gold standard over and above the much neglected physiological stress test.
Of course, the limitation of stress test is that , it has only 75% specificity( to rule out CAD ) and about 80% sensitivity (To detect CAD ) .In simple terms stress test is likely to miss 20% times to miss a CAD in patients with CAD and 25% of times falsely diagnose CAD in patients without CAD.
In the above statistics , coronary angiogram was considered gold standard . The problem with this data is that , CAG is not the real gold standard ,but it was nominated as a gold standard . We now know normal coronary angiogram is not equivalent to normal coronary arteries and vice versa.
While both test have limitations , it is logical to believe CAG has an edge over stress test since it visualises the anatomy. But , once an obstruction is demonstrated by CAG, stress test scores over in assessing the physiological impact of the lesion.
Is a 70% LAD lesion significant or not ?
Stress test will give vital information to answer this question.If this patient performs 10-12Met exercise without symptoms it means , the obstruction is not impeding the flow even during stress. He may do well with medical therapy.
What does a positive stress *mean for the patient and for the physician ?
(* A false positive EST in LVH, anemia, baseline ST shifts are included in discussion )
A positive stress test with or without angina at low workload <5 METS indicates very significant obstructive CAD either in left main , or proximal LAD/LCX. They should getimmediate CAG.
A positive stress test at load 5-10METS is again significant and patients should get early CAG
A positive stress test with angina at good work load >10-12 mets would indicate insignificant or minimally obstructive CAD.
A positive stress test at the peak of exercise at good work load > 10-12METS without angina could indicate a false positive or very minimal CAD.
For the physician , the proper way of interpretation should be , the fact that a person performs 10-12 METS indicate the myoacardial blood flow would be more than adequate in most life situations. Knowing the coronary anatomy serves no purpose here, as no revascularisation will be attempted even if he is going to have a significant CAD ( Which again , is also highly unlikely ) .He should be managed with appropriate lifestyle (Diet, activity, relaxation ) anti anginal drugs, aspirin , good lipid control and plaque stabilisation with statins .
Can a patient with critical left main or proximal LAD perform >10METS in exercise stress test ?
No , large clinical experience (Also refered to Class C evidence by ACC/AHA!) indicate no patient with critical left main or equivalent disease can perform 10 METS excercise
While , EST may be less hyped investigation, but it is the only noninvasive test , ( that too , simple and cheap ) that can rule out * a significant left main or equivalent almost 100% correctly .
Now that, the results of COURAGE and BARI 2D have clearly indicated medical therapy is best form of management in chronic CAD , ( except in severe obstructive CAD in vital locations) a positive EST at > 10-12Mets , has absolutely no indication* to for doing a CAG.
*Some would advocate a policy of doing a CAG as a baseline investigation in all patients with positive EST to know the coronary anatomy and will not proceed onto revascularisation if there is insignificant lesions.
Further , real life experience has taught us , routine CAG in these patients
Increases patient anxiety as he is given a report with a diagram of obstructed heart vessels
Leads to multiple cardiac consultations
Divergence of opinions
Finally end up in the likely hood of a inappropriate revascularisation for a insignificant distal CAD.
Final message
Every patient, who has positive stress test , ( Please note , it could even be true positive ) need not undergo CAG . Most interventional cardiologists could feel otherwise , but one should also remember , There is one more role for the interventional cardiologist ie , to intervene when inappropriate interventions are done to their patients.
NSTEMI constitutes a very heterogeneous population .The cardiac risk can vary between very low to very high . In contrast , STEMI patients carry a high risk for electro mechanical complication including sudden death .They all need immediate treatment either with thrombolysis or PCI to open up the blood vessel and salvage the myocardium.
The above concept , may be true in many situations , but what we fail to recognize is that , STEMI also is a heterogeneous clinico pathological with varying risks and outcome ! Let us see briefly , why this is very important in the management of STEMI
Management of STEMI has undergone great change over the past 50 years and it is the standing example of evidence based coronary care in the modern era ! The mortality , in the early era was around 30-40% . The advent of coronary care units, defibrillators, reduced the mortality to around 10-15% in 1960 /70s . Early use of heparin , aspirin further improved the outcome .The inhospital mortality was greatly reduced to a level of 7-8% in the thrombolytic era. And , then came the interventional approach, namely primary PCI , which is now considered the best form of reperfusion when done early by an experienced team.
Inspite of this wealth of evidence for the superiority of PCI , it is only a fraction of STEMI patients get primary PCI even in some of the well equipped centers ( Could be as low as 15 %)
Why ? this paradox
Primary PCI has struggled to establish itself as a global therapeutic concept for STEMI , even after 20 years of it’s introduction (PAMI trial) . If we attribute , lack of infrastructure , expertise are responsible for this low utility of primary PCI , we are mistaken ! There are so many institutions , at least in developing world , reluctant to do primary PCI for varied reasons.( Affordability , support system , odd hours ,and finally perceived fear of untoward complication !)
Primary PCI may be a great treatment modality , but it comes with a inherent risk related to the procedure.
In fact the early hazard could exceed the potential benefit in many of the low risk STEMI patients !
All STEMI’s are not same , so all does not require same treatment !
Common sense and logic would tell us any medical condition should be risk stratified before applying the management protocol. This will enable us to avoid applying “high risk – high benefit” treatments in low risk patients . It is a great surprise, the cardiology community has extensively researched to risk stratify NSTEMI/UA , it has rarely considered risk stratification of STEMI before starting the treatment.
In this context , it should be emphasized most of the clinical trails on primary PCI do not address the clinical relevance and the differential outcomes in various subsets of STEMI .
Consider the following two cases.
Two young men with STEMI , both present within 3 hours after onset of symptoms
ST elevation in V1 -V6 , 1 , AVL , Low blood pressure , with severe chest pain.
ST elevation in 2 ,3, AVF , hemodynamically stable , with minimal or no discomfort .
In the above example, a small inferior MI by a distal RCA occlusion , and a proximal LAD lesion jeopardising entire anterior wall , both are categorized as STEMI ! Do you want to advocate same treatment for both ? or Will you risk stratify the STEMI and treat individually ? (As we do in NSTEMI !)
Current guidelines , would suggest PCI for both situations. But , logistic , and real world experience would clearly favor thrombolysis for the second patient . Does that mean, the second patient is getting an inferior modality of treatment ?
Not at all . In fact there is a strong case for PCI being inferior in these patients as the risk of the procedure may far outweigh the benefit especially if it is done on a random basis by not so well experienced cath lab team. (Note : Streptokinase or TPA does not vary it’s action , whether given by an ambulance drive or a staff nurse or even a cardiologist ! .In contrast , the infrastructure and expertise have the greatest impact on the success and failure of PCI ) Final message
So , it is argued the world cardiology societies(ACC/ESC etc) need to risk stratify STEMI (Like we do in NSTEMI ) into low risk, intermediate risk and high risk categories and advice primary PCI only for high risk patients.
This is a 15-year-old post about LVH, written in 2008. Few of my colleagues, now agree with this, still hesitate to oblige in the open, suggesting it is too good to be true! Re-posting it for your own assessment. Surprised, why cardiology community didn’t consider this observation worthy to pursue.
Advantages of Left ventricular hypertrophy (LVH)
Left ventricular hypertrophy is one of the most common clinical cardiac entity.It is recognised either by ECG or echocardiography.LVH has a unique place in cardiology as it can imply a grossly pathological state or a marker of healthy heart as in physiological hypertrophy in athletes.
Logic would suggest, in this era of stem cells and nano medicine , every muscle fibre in ventricle is worth in gold !. So when the nature provides an extra reserve of myocardium in the form of LVH one should welcome it , if otherwise not harmful.
Is LVH due to systemic hypertension benign ?
Not really, LVH has been shown to be an independent cardiac risk factor. (The famous Framingham study)Further LVH can result in diastolic dysfunction and the risk of cardiac failure increases.
But in spite of these observations, an astute clinician with considerable experience will appreciate , patients with LVH fare better during an acute coronary syndrome !
This has been a consistent clinical observation . (Shall we call it as class C . ACC /AHA evidence ? )
Is LVH an asset during ACS ?
A hypertrophied heart takes ischemic injury very easy , it doesn’t really hurt much . Another possibility is that in LVH myocytes are relatively resistant to hypoxia .
Patients with LVH rarely show significant wall motion defect following an STEMI.This is probably because the full thickness transmural necrosis is almost never possible even if extensive MI occurs.
This is also reflected in ECG as these patients rarely develop q waves in following STEMI .
Persistent ST elevation and failed thrombolysis is very uncommon in pateints with LVH.
LVH provides a relative immunity against development of cardiogenic shock . It requires 40% of LV mass destruction to produce cardiogenic shock.This can rarely happen in LVH. In a long term analysis we have found none of the patient with LVH developed cardiogenic shock following STEMI.
LVH patients are also protected against development of free wall rupture.
Concluding message
“Lack of published evidence is the weakest evidence to dismiss a true myth”
LVH , either pathological or physiological, has a hitherto unreported beneficial effect.It acts as a myocardial reserve and helps limit the impact of STEMI.
As the medical literature expands exponentially, the quality and intent of the research questions sound awry. There are only a handful of journals like JAMA that are bold enough to ask some tough and pragmatic questions in this glitzy world of medical extravaganza.
The current issue wants to set the pace for an important debate, on a topic that is rarely discussed.
Check whether your answers concur with this crucial query from Harvard Medical School and Massachusetts General Hospital. Three questions this article wishes to address.
1.What is the reason it is happening?
2. What are the implications?
3. What can be done for it?
My thoughts
“It is indeed overdiagnosed. Once labeled, a chain reaction is set in. The cost, and resource consumption that follow a misdiagnosis are nearly identical to that of a true MI. More than that, the adversities of the tense investigative protocol can convert a misdiagnosis into a real one because that sadly includes even an overzealous poking right at the mouth of the coronary artery just o exclude a non existing MI . and ICU-related anxiety stand apart in this scientific comical game of ruling out a cardiac emergency.
The paper seems to blame mostly on the powerful screening test high sensitivity Troponin, Everyone will agree it has a major role in this. But, the more important reason is the cardiology community’s vigorous adoption of a universal definition of MI criteria (which is never intended to apply at the bedside) .Next factor is probably more important. The fear of missing a potential MI and legal consequences thereafter. I wish, the experts who sit on medical juries need to learn few extra lessons in the art of medical uncertainties.
Medical jurists, need to take some Intellectual cues from their criminal courts. How is it that, even well-planned criminal murders are successfully allowed to be argued and won in courts,…while inadvertent events such as missing aninconsequential MI by doctors are rarely pardoned?
How to avoid over diagnosis of MI ?
In this scenario, It is sad, that only very few cardiologists have the guts to ignore this omnipotent molecular sub-fraction of cardiac muscle Troponin, with their clinical skills. What we can do, at our level is to incorporate a new term “benign or micro myocardial Infarction” – akin to lacunar infarcts or TIA equivalents of the brain in the heart. We need to de-list the vast majority of chronic ischemic,non-ischemic, or systemic causes of Troponin leaks from the myocardial infarction chart. Physicians must realize, that protocol violation should not be deemed a crime always, rather it has a sure potential to benefit your patient if it is done properly and intelligently.
Final message
Maybe, this ranks top among all controversial statements in my 15 years of writing. But, this time it is not self slandering my own profession. Doctors should be legally allowed, (rather forgiven) to make permissible levels of errors in the medical decision-making process . This should include overlooking apparently positive lab results if they have reasonably applied their clinical acumen. *Until this happens, the unquantifiable suffering of our patients due to over-diagnosis and inappropriate interventions can not be reigned in.
I thought, it was pacemaker extrusion. It was indeed a close answer, still terribly wrong. It is an intentional exterior placement of a permanent pacemaker generator mimicking an extrusion due to pocket infection. Here is a patient, where a permanent pacemaker was kept temporarily for a few weeks or a month in high-risk reversible complete heart block situations. This typically occurs after an inferior posterior myocardial infarction, drug-induced CHB.
Currently, with the arrrival of TAVR, CHB has beceome a glamorous complication and is getting wider attention. This happens due to the anatomical uncertainties where the inferior landing zone of TAVI is pre-destained and is beyond our control. This is more true in the self expanding Core valve platform . When the lower edge treaspass the non-coronary cusp- membranous septal junction, it hits perfectly the compact post-penetrating bundle of His, confering a high risk of CHB.
Still, the good thing is some of them recover as the pressure edema regress .Putting a PPM in all such patients was considered mandatory or even a vanity in the past. Now we realise it is an additional metallic luggage in an already strained heart, Temporary-PPM the oxymoronic innovation is perfect option in this setting.
Final message
A typical external temporary pacemaker can be kept for up to 2 weeks maximum. (We have kept it for a month or so) It’s done via the jugular, subclavian, or even femoral. If the underlying condition demands more time for recovery of CHB, many do a regular permanent pacemaker.
Now , we have this unique option of using PPM as TPM. This is not a new concept though. It was used few decades ago .Has come back in more centers .Thanks to TAVI and its specific complications.
Reference
1.Rodés-Cabau J. Ellenbogen K.A. Krahn A.D. et al. Management of conduction disturbances associated with transcatheter aortic valve replacement: JACC Scientific Expert Panel. J Am Coll Cardiol. 2019; 74: 1086-1106.
2. Leong D, Sovari AA, Ehdaie A, Chakravarty T, Liu Q, Jilaihawi H, Makkar R, Wang X, Cingolani E, Shehata M. Permanent-temporary pacemakers in the management of patients with conduction abnormalities after transcatheter aortic valve replacement. J Interv Card Electrophysiol. 2018 Jun;52(1):111-116. doi: 10.1007/s10840-018-0345-z. Epub 2018 Mar 12. PMID: 29532275.
Lowering the raised LA mean pressure is a major therapeutic goal in any severely symptomatic left heart disease, whether it is valvular or myocardial disease. It is prudent to understand, that even in systolic LV failure; it is the raised LVEDP that causes the symptoms and marks the limits of exercise capacity. Drugs like inotropes, pre-load , afterload modulators like diuretics and vasodilators can take care to a certain extent.
When symptoms are refractory and the underlying condition has no primary correction , we need to intervene with some extreme procedures. We know a small ASD decompresses mitral stenosis, and the combination of ASD and MS, Lutembacher, is a well-known syndrome called Lutembacher. The concept of LA flow regulator or decompressor came from this .
When the left ventricle is stiffened and restrictive, and LA mean pressure is prohibitively high,we have a viable option now. This is to create a small regulatory orifice in the IAS ( A complicated term for a small ASD) to decompress the LA and reduce pulmonary congestive symptoms. Curious minds might ask, can’t we decompress LV it self by creating a small VSD. Probably in the thin membranous area. May be, it will come soon in the innovative lanes of cardiology.
The study finds the device can be beneficial without compromising much on RV side function.
Animation Courtesy: Corvia website; The procedure looks simple when compared with other procedures inside the LA .The device looks like an octopus, and sits on either side of IAS, like a stapler and maintains the orifice.
Here is an audio podcast from the primary author published in JAMA network.
Interview with Sanjiv J. Shah, author of Atrial Shunt Device Effects on Cardiac Structure and Function in Heart Failure With Preserved Ejection Fraction: The REDUCE LAP-HF II Randomized Clinical Trial. Hosted by James E. Udelson,
Final message
This device’s core concept lies in requesting the right ventricle to help its bigger brother LV at its difficult times. You can call this an artificially created interventricular dependence. Though it might help, we need to watch the right heart’s dynamics closely. Maybe, if RV is experiencing difficulty, we can have external control over the IAS orifice and flow as and when required. (This is not new, a remote-controlled switch regulation was done for pulmonary banding in children with congenital heart disease who needed regulation of pulmonary flow by a device FloWatch-R-PAB (Ref 2)
It is logical to expect the same device would be useful to decompress RA at high pressures as in severe primary pulmonary hypertension. If you think backwards, it looks the same as a life-saving, reverse Rashkind procedure variant in adult
Primary PCI of IRA , continues to be a clinically popular & statistically validated (In spite of some critical ifs & buts) coronary reperfusion strategy.
What to do, if we happen to detect, a significant or borderline lesion in the Non- IRA territory during pPCI ?
There are too many guidelines scattered across cardiology literature to “help or confuse” us on this issue. They argue for either immediate intervention or defer transiently, postpone or just ignore , based on clinical ,hemodynamic*, Individual, institutional , or some other non academic factors. (Permanently deferred PCI is other wise called medical management, is practiced by some inferior cardiologists or GPs who never refer such patients to higher centers after a stand alone thrombolysis)
* The FFR, iFR RFR, related stuff
What if if we are completely blinded to the status of Non IRA vessel ?
What do you mean ?
I mean , can we be, “not- aware” of contra-lateral lesion status ?
Yes, “Simply don’t do a non IRA angiogram , that’s it. If its RCA PCI , don’t shoot Left main, and vice versa for LAD. Do a PCI without doing a completed CAG. I mean IRA PCI alone, by guessing it by ECG .
What a crazy Idea ?
This week’s JAMA has something* relatable to this idea. The aim was to do PCI before complete CAG , to document any advantage. (It is important to note, CAG was done in all patients)
Did this study really happen ? Seem to have many ethical issues . That too, published in JAMA net work. Yes, it was done, I guess , with a legal protection . Apparently, It was done without an informed consent even.
Was there any advantage of proceeding directly to IRA PCI ?
Yes. Reperfusion times were significantly shorter as expected.
Any other advantage ?
I think there are definitely few more, that can’t be reported by many of us, for either unscientific or ethical reasons.
Any disadvantage?
Proceeding to do PCI without knowing contralateral coronary status is unprofessional act and potential to end up in low quality reperfusion.
Final message
Incidentally, this study raises lots of interesting possibilities. Why should we know , the status of non IRA at all, if IRA is opened and flowing well ?( Missing a critical lesion in non IRA is a crime , is it not ?) I agree. but, don’t use big words. Wish some one does a study with totally blinded about non IRA status, however unethical and unscientific it may be. After all, globally 90% of all successful myocardial reperfusion is done by the humble streptokinase or the more glamorous TNK -TPA . Both these agents never bother to know, which coronary arterial thrombosis its going to work .
As a learnt cardiologist, we must realize most of the STEMIs can be managed successfully* without even knowing which is the IRA, forget about the non-IRA. Tackling non IRA lesion is never considered as an emergency procedure , in fact it carries a fair chance of precipitating one.
*Post-amble
Beware, the conclusions of this study, and the core suggestion in this post may-not be related , if any one finds , it is at their own cost of “whims or wisdom“
A 76-year-old woman with a history of double valve replacement (Aortic and mitral valves) for rheumatic heart disease, presented with acute dyspnea after a switch from Warfarin to LMWH before a planned bone marrow biopsy.
The investigations revealed a stuck aortic prosthetic valve ,that showed a prohibitive gradient of more than 50 mmhg. Since, she refused further surgery, a rare and risky effort was made to balloon dilate the prosthetic valve leaflet, though it is not a standard approved modality. It was decided to dilate the supero-lateral orifice and the central orifices by simultaneous kissing balloon. The results were dramatic.
The images and video are reproduced with courtesy of Dr. David Smith, Dr. Ayush Khurana, Department of Cardiology & Cardiac Surgery, Morriston Cardiac Centre, Swansea Bay University Health Board, Swansea, United Kingdom
The stuck valve
Twin balloon dilatation of bi-leaflet valve in between the superior and central orifice
There are few important lessons from this rare case report.
The innovative double balloon catheter Inflation across the the mechanical prosthetic valve is possible. This technique is likely to emerge more useful in the post TAVI population as well.(JSCCAI 2023)
Some times, a simple maneuvers like tapping , pushing or releasing stuck leaflet will solve the issue in few lucky patients. The reason is a clot less than 2mm can strategically sit on the hinge point and interfere with its motion. Dislodging a 2mm clot in all likely hood cause a benign TIA , or just vanish in the aortic stream down the hill,
However , the risk of thromboembolism is genuine in those a clear thrombus is visualised. Hence distal protection by an Aortic sentinel device or its equivalent (FilterWire EZ, Tri-guard) is a must. If Aortic protection device is not available, proceeding with patient & family consent is not forbidden if circumstances demand.(In India ,we do PTMC with mini LA clots without any protection) A video on Sentinel aortic filter
4.It is to be noted if the obstruction is due to pannus , risk of thrombosis is almost nil and safety of prosthetic balloon valvuloplasty is almost ensured.(Of course with risk of device leaflet damage )
5.As on today, differentiating pannus from thrombus remains continues to be a learnt clinical guess game. CT and MRI can give more crucial inputs. To make things more difficult , a raw area over pannus could be the nidus for the thrombus.
6.Probably , the major learning point (rather a sort of mistake) is the decision to switch over to LMWH in lieu of OAC. Time and again we have seen LMWH is a weak anticoagulant, with erratic correlation of Anti X-a activity and efficacy.
7.I believe, in the above case. this complication might not have occurred if she had continued on OAC , if that was not possible , a switch to regular un-fractioned Heparin as a bridge during the surgery could have been the right choice. Generally, overestimation risk of bleeding viz a viz with life threatening thrombosis is quiet common especially in patients with prosthetic valve.
Current approach for prosthetic valve obstruction
A comprehensive review and surprise inclusion of leaflet release as an option.(Ref3)
The well known pro-coagulant state of pregnancy is an evolutionary protective process to make blood clot quicker, to save fetal loss in early pregnancy and mitigate postpartum bleeding. Still, in many women, this natural adaptive process confers an enhanced thrombotic risk. The molecular mechanisms for this pro-coagulant state are, there is increased factor VII, fibrinogen, reduced protein S. It is interesting to note, while plasminogen levels are elevated, D-dimer is also increased, indicating an ongoing fight between pro & anticoagulant forces, converting the physiological maternal- placental bed a mini harmless DIC equivalent zone.
There are several important systemic, placental, (Fetal) and cardiac indications for anticoagulants and antiplatelet agents in pregnancy. The list is increasing in a steady fashion. (Most IVF pregnancies seem to need it for some unknown reason)
Finally most importantly prosthetic heart valves & other Intra cardiac devices.
We have few options
Warfarin (Molecular weight 300 Daltons) is used in dose of 2- 10mg
Un-fractioned regular Heparin , (40000 Daltons) -Not practical for long term. Used at peripartum phase , just before labor to take control over possible PPH.
LMWH (Molecular weight 5000 Daltons)
NOACs are not an option as of now
Aspirin alone might give partial or near complete protection in some of the above mentioned indication.
General rules
Warfarin is safe for mother, Heparin is safe for fetus .(both Un-fractioned heparin & LMWH )
Just because heparin is safe, we cant choose t, it must be equally efficacious too. (Till date no study on LMWH has come to show its efficacy any where closer to Warfarin efficacy, (forget about beating it) in protecting mechanical valve events)
The concept of bridging till 12 weeks is not mandatory in all
Switching to regular heparin at term is applicable for both
Any anti-coagulant usage in pregnancy is like playing with fire .They have narrow safety window. Further, we must have have a quick antidote in case of dose excess. Warfarin, a powerful VKA, is the time tested key drug despite the well known teratogenic effect. Now we have an alternative LMWH ,which has gained considerable popularity.
The risk of Teratogenicity in warfarin is absolute or is it dose dependent ?
Yes it is dose dependent. (Warfarin causes two phases of side effects one is embryopathy, it also affects later half of pregnancy ie fetopathy with neurological bleeding etc
The Italian connect
Answer to this question came from oldest Romanian city built by the Greeks, Naples, Italy . Dr.Vitale , from the department of Cardiac Surgery, Monaldi Hospital, did this landmark study, way back in 1999 , and convincingly proved , the dreaded embryological side effects are dose dependent. It was done with a meager 58 pregnant women . The conclusions of the study changed the way we used to worry about this drug. It said, warfarin is safe at low doses even in the first trimester , if used <5mg, in terms of embryo and fetal issues. Isn’t it curious that a dreaded drug was made pregnancy friendly by simple study from smart surgeon . It is a real surprise that the conclusion of this study is still can’t be disputed by another big one. Almost all current guidelines use this 25-year-old study to form the core algorithm of current anticoagulant protocol in pregnancy.
Warfarin vs LMWH debate
Teratogen or No-teratogen, coumadin still rules supreme in most high risk situations, especially in women with mechanical valves, (Despite the ease with which this molecule crosses the placental filter , because of low molecular weight -300)
Heparin one of miraculous drug of last century , remains a life saving anticoagulant for various medical conditions. However, its refined version LMWH, though made it more palatable & user friendly, it un-apologetically took the sting out of regular heparin, made it less efficacious (more glamorous though) LMWH usage is in CAD widespread , it has suspect value* in true ongoing ischemia in any active ACS situation. It is strange anti X-a is never monitored in CAD protocol , while in pregnancy we insist on intensive monitoring i. What does it imply ? Monitoring is primarily done to ensure adequacy of anticoagulant activity , rather than risk of bleeding .
In my 30 years I am yet o see a patient have fatal bleed to poorly monitored Enoxaparin. This is the reason the mid trimester LMWH heparin bridge to collapse in many pregnancy anti-coagulant protocols. Now ,we can understand why the veteran VKAs continues to be a flag bearing drug in pregnancy .Of course, INR-guided OAC therapy, though can be tricky, most of us are used to that, unlike the frightful anti X-a troughs and peaks.
*I am sure most Interventional cardiologists will hesitate to disagree with this observation.
2022 update on LMWH : More trouble for LMWH in pregnancy. There considerable concern , that twice a day sub-cutaneous injection may not maintain target anti -X a trough .6-.8U/ml and peak 1 to 1.2 U/ml and currently many centers advice LMWH three times a day ( Bai C, Wu . Medicine (Baltimore). 2022 Dec 30;101(52):e32550.)
Final message
So far, the traditional dictum has been, Warfarin is safe for mother & heparin is safe for fetus. One has to decide accordingly with patient ,spouse & family. I think, its time to tweak this rule, little bit. Warfarin is safe for both mother & fetus* in most patients till 36 weeks in low doses , while LMWH may be more safe , but lags far behind in efficacy, especially in high risk indication. (*Including first trimester but with a lesser proof though)
Postamble
Even in these era of shared decision making, it’s our duty to impress upon our patients (or even enforce) to choose warfarin over LMWH in appropriate times. Don’t simply leave this critical decision to patients.
I think we need another study ,5, 8 and 10 Warfarin vs LMWH with a prosthetic valve and analyze the fetal bleeding risk in mechanical valves. It may not be a surprise if the cut off of 5mg could move further up.
I don’t know, whether it is a good trend, to note more and more biological vales are implanted at an young age to avoid OAC .These valves have short life span demanding redo surgeries within 10-15 years which may not be not a righteous approach.
A 32-year-old high-profile businessman was advised Holter monitoring for a few ectopic beats during routine screening ECG. The 72-hour extended Holter monitoring picked up a single short pause with a blocked P wave and reported as doubtful Mobitz type 2 AV block.
The cardiologist in-charge, told the patient that findings are significant, and he would need further investigation. He was referred to their associate center for an EP study. After hearing about the procedure ,the patient was freighted about inserting multiple catheters inside his heart.
This was the time he consulted me with Holter report. It was indeed a missed QRS after a well inscribed p wave , recorded at 4.57 AM, It is a 2nd degree AV block, may be Mobitz type 2, . What if ? It could still be be blocked atrial ectopic. (Pseudo AV block) Both preceding and following PR intervals seemed to be non varying . The following QRS was narrow. I don’t know, whether a single blocked P could by any way a concealed Wenke -Bach. I didn’t have calipers to measure the PR accurately though. The baseline heart rate was around a vago-genic 60/mt, that was comforting . He had his echocardiogram done already and was normal.
What does the guidelines say ?
Guidelines are short of evidence , it was as vague as my thought process . It suggested EP study in selected patents with asymptomatic second degree AV block . My fellows tell, it is just 2B indication (To-be frank, 2-B indications should be called as a junk recommendation ) which would mean if you wish you can do a “potential harm”
I asked the patient two questions.
1.Does he have any symptoms like dizziness or syncope ?
Absolutely nil.
2.What is his functional capacity?
Excellent.
That’s great. Within a minute or so , I could confidently confirm, the non-seriousness of the Holter tracing. I asked him to forget everything, and sent him home, with reassurance, taking on myself a miniscule risk of missing a true AV block and its consequences. He thanked me profusely with so much gratitude. Every thing was hunky-dory , then , this thing happened. When he was above to leave the office, he came back. “Doctor, I forgot to tell, my father died suddenly at the age of 48 apparently by a heart attack” .I must admit, I was taken aback the moment he told this.
What an important past history, I failed to elicit earlier. As he left my room, I called my secretary to give a Suo-moto appointment to him 2 weeks later with a plan of TMT and possible CT -angiogram. Till late in the evening, this patient’s Holter recording ran in my mind. What was that reason for original VPDs that invited a Holter test and the subsequent documentation of Innocent appearing AV block ? Are they interconnected or inherited ? or Is it really Ischemic ones, that took his dad’s life?
That was enough for me to make a compelling call to my EP colleague, for a quick chat about this unique patient. We discussed for 15 minutes, right from Padua University paper to all the Brugada variants.(Ref 3) In the end, the basic doubts remained as before. However, the patient was advised for an EP study primarily to know the HV interval and the possibility of diffuse distal disease. The possible need for a MRI study to rule out silent arrhythmogenic intramural granulomas was also discussed. My EP friend poked me with more academic toxemia. He said a screening test called cardiac-arrhythmic genome analysis is available in certain European centers. Ref: Isbister, J.C., Semsarian, C. The role of the molecular autopsy in sudden cardiac death in young individuals. Nat Rev Cardiol21, 215–216 (2024).
I said enough is enough , and requested for hanging up the chat.
Final message
AV blocks, even Mobitz type 2, can occur at normal times of heightened vagal tone.(Massie Block-Ref 1) But, if there is something unusual in the clinical history, be ready to investigate until the arrhythmia, or at least the anxiety disappears.
Opening a CTO, for reasons other than angina (i.e. for relief of dyspnea or improving functional capacity) is largely conjectural and based on randomly accrued data backed by poor interpretation. The role of collateral circulation in CTO that can compensate even during exercise is well known at patient level data. This has become a difficult area of research because it involves spending more time with the patient, and hence not studied much. We are in the era of artificial intelligence ,virtual patients and statistical extrapolations that can steer the Kaplan Meyer curves in the desired direction.
Pure academicians shall follow the current guidelines. Surprise… surprise !, There is some good news. The normally aggressive American guidelines exercise much caution with a 2B punch. Still , even today it is weird to see hours of academic time is consumed in CTO Interventions in any interventional cardiology meets. (May be , they could get a breakthrough benefit , which I couldn’t appreciate)
.
CTO-PCI follow up
The incidence of MACE including ACS varies between 12-28% depending on LV function.(Ref 2) How about Conferring 12% risk of ACS in a person who has normal LV by doing CTO-PCI ? Still it continue to be a smart move for many of us ? This is exactly the reason experts are struggling to come to term with truths behind negativity of most published CTO trials.
Now, answer to the title question. What is the future risk of ACS in opening CTO related artery ?
Asymptomatic CTOs, with fair excercise capacity, should probably never be opened for the simple reason, a closed artery is naturally protected, against a future ACS at least in its territory
Final message
Currently, in the science of cardiac revascularization there is only evidence and it’s Interpretations, little patient level facts.
Living with a single coronary artery, is potentially a frightening scenario for the patient* which has to supply its own area and also, need to donate the occluded coronary artery . What will happen if a single donor (RCA/LCX) gets closed? One more remote risk in CTO is, acute collateral shutdown causing STEMI/NSTEMI. These statistically minuscule risks are well exploited by coronary caretakers. Meanwhile, there is little talk about the chances of CTO getting closed by itself after an apparently successful PCI. The consequences of anatomic and hemodynamic collapse of hitherto well flowing collaterals , after a CTO PCI will require a separate discussion.
*It is wiser to recall , left coronary artery is also single before bifurcating. Surviving an entire life span with a single10-20 mm tunnel called left main, rarely elicit the same fear in us.
It is a120-year challenge. Can anyone replace Rontgen’s X-ray discovered in 1895 for medical imaging? The Nobel winning Invention redefined the way we looked at our body and management of diseases for over a century. However, the fact remained it is an invasive and injuring investigation. What is the alternative for the X-radiation ?
CT scan was a great invention, but it turned out to be a gigantic 360-degree clone of X-ray machine. Today’s cath lab, however sophisticated , is like spending hours together inside a hot Chernobyl coffee shop. MRI was a true game changer. With zero radiation, MRI came close in the fight with innocuous proton imaging. But for live cardiac interventions, MRI was not practical. Meanwhile, over the years, ultrasound moved up from the pelvis, abdomen, right into coronary arteries and heart. Intravascular ultrasound-based interventions are being done in coronary artery, in a few cases to avoid contrast in patients with CKD. (We call it zero-contrast IVUS-guided PCI). But, it is cumbersome and has some technical issues. Transesophageal echo (TEE) & Intracardiac echocardiography (ICE) do help us immensely in certain interventions.
Now is the era of Optics
If a torch light can illuminate and give us vision in absolute darkness ,how about acquiring a deep vision with scattered light ie photons. (Jnana-Chakshush ,third eye of Hindu God Shiva ?) The concept of Optical coherence imaging came (OCT) came in .It has limited use in deep vision of coronary wall anatomy and histology. As of now it has no role to play in catheter guidance.
Here comes the real Innovation . Fibro-Optic real shape( FORS) technology , which reconstructs image from optical data, and beam live fluro- like images, in 3 dimension. May be, we may soon, say good bye to electrons, protons, and welcome these harmless photons.
This video clip shows real time Intervention using FORS in Aortic endovascular stenting
One may Imagine FORS to Electro-physiologist’s electro- anatomic mapping made with a the GPS like pad attached beneath the cath table and reconstructing anatomical Images from the dynamic signals points generated from the catheter tip.
Final message
Now, we are looking at various different modalities to image without radiation injury to the patients, and more importantly the cardiologists .
Intra cardiac Echocardiography (ICE)
Proton imaging (MRI)
Electrical navigation (CARTO)
Fibro-Optic real shape (FORS)
FORS , is the new arrival. Let us hope it stands the test of time.
Click below to see who is watching this website live !
This site will never aim for profit. Still ,this donation link is added at the request of few visitors who wanted to contribute and of-course that will help make it sustainable .
Please Note
The author acknowledges all the queries posted by the readers and wishes to answer them .Due to logistic reasons only few could be responded. Inconvenience caused is regretted.